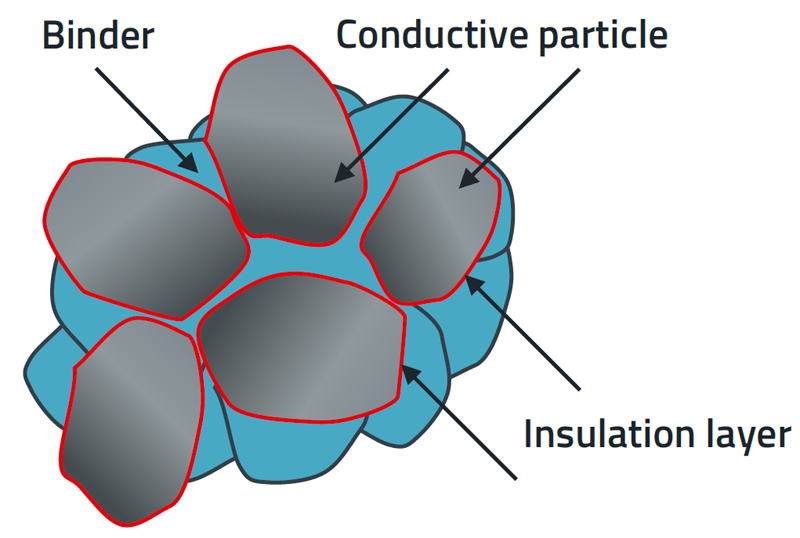
Quality management system meets the demands of the medical device market
When companies are certified by international standards or qualified by a regulatory body, it signals to customers that these companies are dedicated to producing a quality product consistently and according to customers' and regulatory authorities' requirements. ISO 9000, ISO 13485 and the U.S. Food and Drug Administration (FDA) Quality System Regulation (QSR) give companies a minimal set of standards with maximum flexibility. They provide guidelines that tell them what to do, without telling them exactly how to do it. Each standard serves a different purpose, all working towards a common objective. ISO 9000, ISO 13485 and QSR all directly affect how electronic component suppliers effectively and successfully sell to the medical device original equipment manufacturers (OEMs).
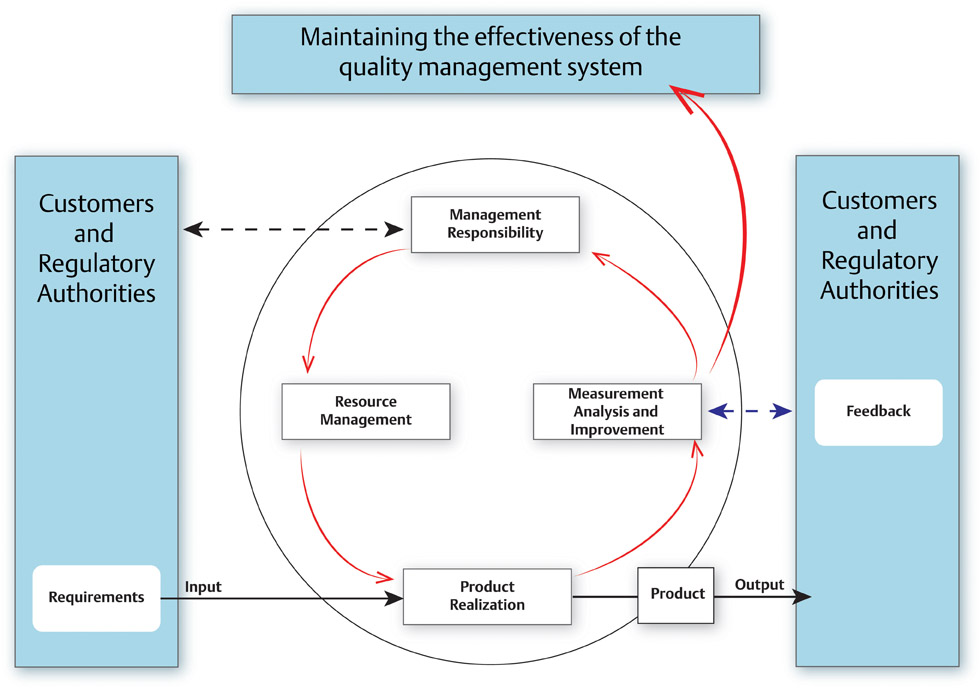
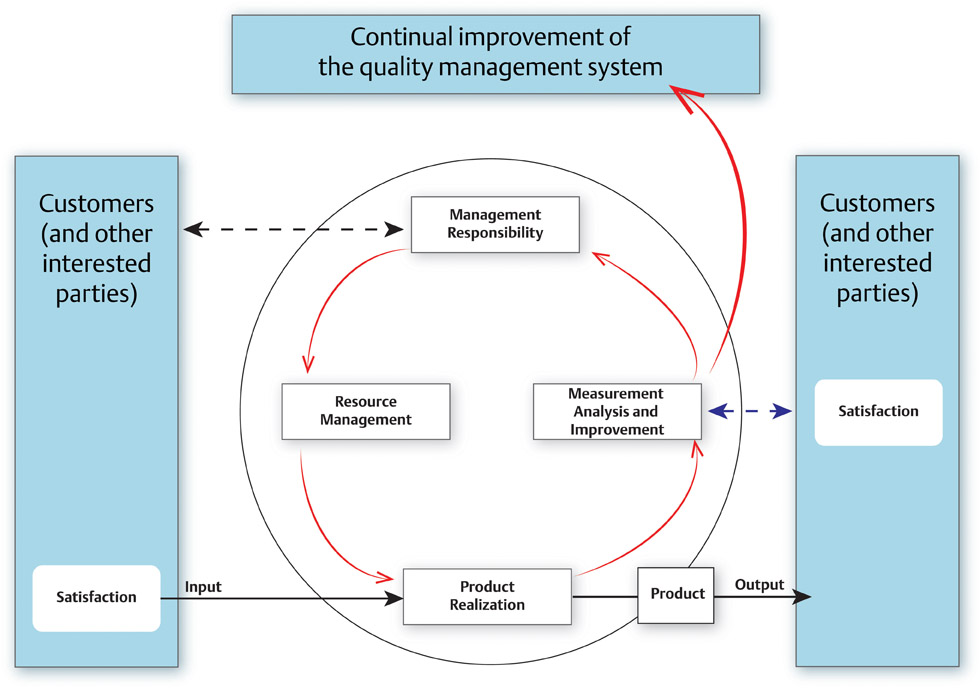
History and Purpose of Quality Management Systems ISO 9000 was first released in 1987 by the International Organization for Standardization, originating from several existing international standards including US MIL-Q-9858, Canadian Standards Association Z299 and British Standard 5750. At the most basic level, ISO 9000 is a quality management system for the delivery of products or services. It provides the infrastructure that fosters effective, efficient and continual change to world-class operations. It is not specific to any industry and had major revisions in 1994 and in 2000. The ISO 9000:1994 revision had 20 elements, covering separate areas involved in running the entire business. In the ISO 9000:2000 revision, the 20 elements were organized into 5 key subsystems with an added emphasis on customer satisfaction and continuous improvement. This 2000 revision highlights the ISO process approach that shows the interrelationships of business processes, feedback loops and trending metrics for continual improvement. In 1996, ISO 13485 was released as a quality management system adapted from the ISO 9000:1994 revision and was established specifically for the manufacture of medical devices. After ISO 9000:2000 was released, the ISO 13485:2003 process version was published. Besides meeting customer satisfaction, ISO 13485 aims to meet the requirements of national health authorities who regulate medical devices sold in their location. It also aims to maintain the effectiveness of the quality management system, not strive for continual improvement. This intends to avoid changes that may affect the medical device safety and essential performance as originally approved. ISO 13485 also incorporates risk management (particularly targeted at safety risk) to decrease the likelihood that a medical device may cause serious injury or death.
To assure the safety and efficacy of products like food, drugs, biologics and devices, the U.S. FDA requires that the manufacture and marketing of such products be made using current good manufacturing practices (cGMP). cGMP places the burden of proof on the manufacturer to ensure that its product and the process used to make it are consistent and controlled. In 1976, cGMP for medical devices was defined in the Medical Device Amendment Act. In 1996, cGMP for medical devices was revised, harmonized with ISO 9000:1994 and ISO 13485:1996, and given a new name: Quality System Regulation, or QSR. QSR officially went into effect in June 1997. Differences between ISO 9000, ISO 13485 and QSR While working towards one goal, each standard and regulation has distinguishing factors that separate it from the rest. The objective for each standard is a major differentiator. ISO 9000 applies to any provider of a service or product. ISO 13485 has a more specific application: manufacturers of medical devices. QSR also applies to medical device OEMs but only those marketing in the U.S. For ISO 9000, the quality management system is focused on optimizing the interconnected processes that bring value to the company and customers and is geared toward continually improving these processes using metrics and feedback. ISO 13485 and FDA QSR have similar foundations but keep sacred the same level of product safety and essential performance as originally approved by respective regulatory bodies. ISO 9000 is different from ISO 13485 and FDA QSR in the same manner as a consumer product like an MP3 player is different from a medical device like a glucose meter. When the MP3 player malfunctions, the consumer simply goes back to the store for a refund, repair or replacement. When the glucose meter malfunctions, it can severely impact a person's health or potentially lead to death. The patient reports it to the doctor and it gets recorded as an adverse event with the FDA or through some other vigilance reporting to the national health authority. Product failure of a non-medical device is an issue of warranties and unsatisfied customers. For medical devices, product failure is an issue of public welfare and investigation of post-market surveillance or vigilance reports. Both ISO 9000 and ISO 13485 are voluntary certifications undertaken by manufacturers on their own money and schedule. QSR, on the other hand, is a mandatory requirement, validated by the FDA or Accredited Person through inspections. Non-compliance to ISO leads to denial or repeal of certification. Non-compliance to QSR leads to administrative and/or judicial enforcement. Inadequate cGMP means adulterated products and can result in actions ranging from a simple warning letter to a major recall or injunction. ISO certifications are like practice review tests that companies take and correct themselves until they get the answers right. FDA inspections for QSR are the actual tests where they pass or fail. Influence on Electronic Component Suppliers Though electronic component suppliers are selected by medical device OEMs based on their components' application specifications (e.g., IEC 60601 medical safety certification), most suppliers are not covered by medical device regulation as regards quality management systems. Not surprisingly, a major cause for medical device recalls comes from component issues. Most suppliers are only ISO 9000 certified. Below are the main elements of ISO 9000, ISO 13485 and FDA QSR where there are differences in implementation. Electronic component suppliers can review these elements and upgrade them to the level demanded by industry regulations to bring more value to their medical customers. On the other hand, medical device OEMs can assess their suppliers' quality systems in these areas and work with them to fill gaps in regulatory compliance.
- Purchasing Control - The three standards are very similar, with ISO 13485 and QSR taking their lead from ISO 9000. However, ISO 13485 and QSR require verification not only on products purchased from suppliers but also on services received. This is important when such supplier services affect the manufacturing process that crucially determines the quality of the end medical device. QSR also requires that suppliers notify medical device OEMs of changes. When informing their medical customers, suppliers can readily do risk assessment on changes on their components as they impact the end device safety and performance.
- Product Identification and Traceability - ISO 9000 is more relaxed and may allow exclusions, while ISO 13485 and QSR are strict, especially on non-conforming products and implantable medical devices. Both standards require records of environmental conditions (e.g., sterility) as well as distributors' records of devices' whereabouts. This is particularly important when suppliers are providing not merely single components but subassemblies. In case of product failures, identifying the specific component, cause of failure and affected lots is critical in carrying out a device recall.
- Design Control - ISO 13485 and QSR require documented procedures for design and development, including risk management with substantiating records. QSR specifically refers to the Design History File (DHF). The DHF is a compilation of records demonstrating that the design was developed in accordance with the approved design plan. ISO 9000 does not have such requirements. Electronic suppliers whose components directly affect the safety certification (IEC 60601-1) must clearly be linked to the DHF.
- Process Control - ISO 13485 and QSR go beyond ISO 9000 particularly in ensuring validation of processes or changes that may affect the medical device safety and performance. Both standards include provisions for cleanliness of product and contamination control. Also, QSR cites validation even for software changes.
- Corrective and Preventive Action - Both ISO standards appear similar in regard to corrective and preventive action; however, ISO 13485 and QSR incorporate analyzing quality data and trends from post-market surveillance data, not just from internal measurements. Oftentimes, this requires rigorous root cause analysis and clear preventive actions to such field failures. Based on post-market surveillance regulations in each country, medical device OEMs have a very limited time to inform the respective regulatory bodies of any adverse event and its resolution.